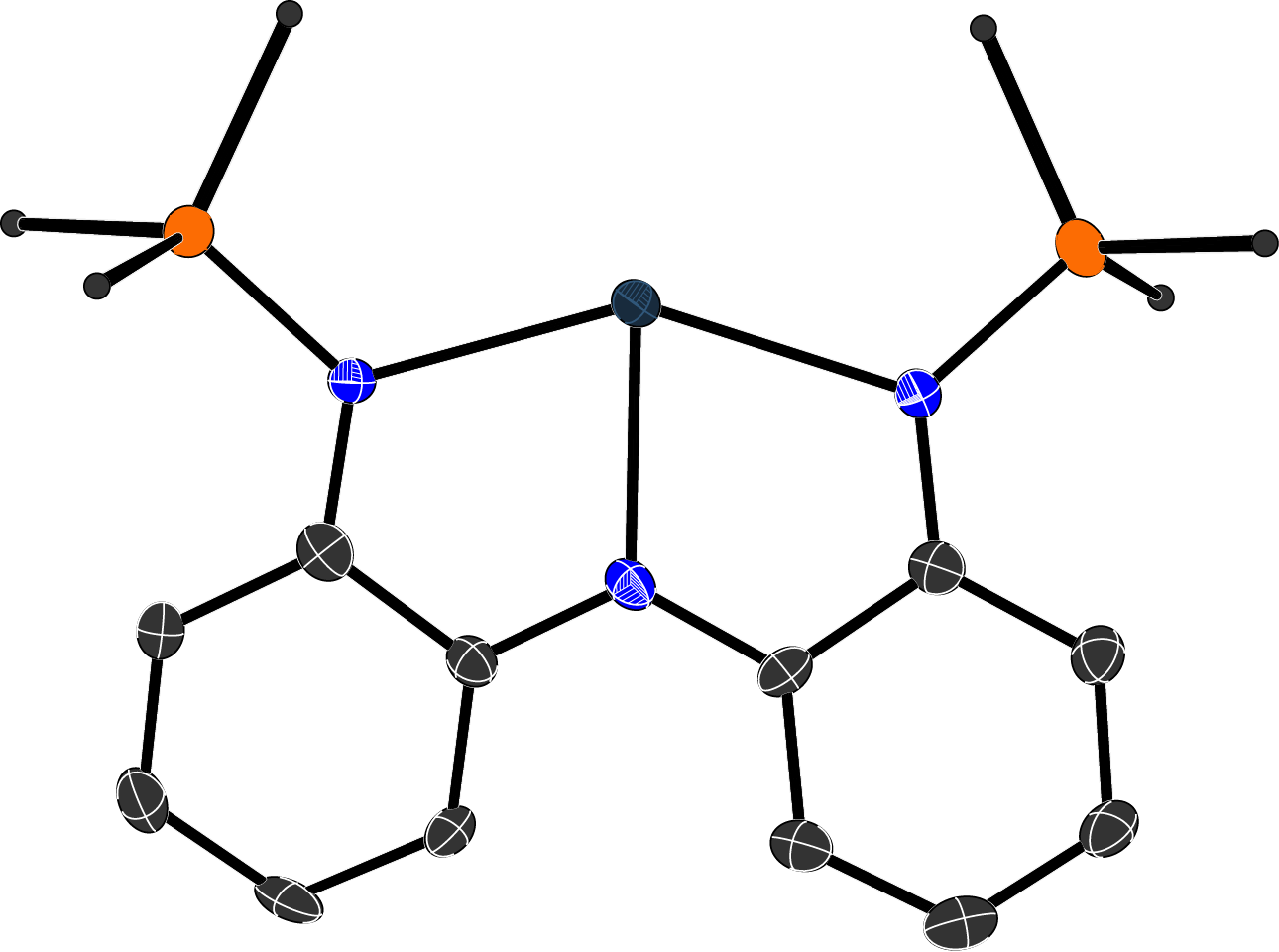
Inorganic polymers & Materials
Synthetic materials are an integral part of modern life as commodity plastics, coatings, fabrics, adhesives, elastomers, foams, and packaging. Most materials have carbon-rich backbones due to the availability of precursors (monomers) from fossil fuels, the strength of C-C, C-N, and C-O bonds, and centuries of organic chemistry knowledge to guide development of new monomers or polymerization methods involving carbon. There are, however, several limitations associated with the use of such organic polymers. First, they maintain our reliance upon fossil fuels for material precursors. Second, they do not readily degrade in the biosphere, resulting in environmental accumulation. Third, hydrocarbon polymers are not suitable in extreme conditions (e.g. growing aerospace industry) due to their thermal, oxidative and radiation sensitivity. Finally, such polymers are limited to the properties of light elements (C, H, N, O), and cannot access the exciting ones available to heavy elements (e.g. neutron or UV-capture cross-section, high nuclear spin, magnetism, spin-orbit coupling, relativistic effects).
We are developing inorganic rings and cages as monomers that can introduce new properties into polymers or materials.
Depending upon the inorganic elements employed, the materials synthesized in this project have a applications in agriculture, biomedicine, nanolithography, high temperature devices, and as stimuli-responsive materials or coatings.
Lead References:
Adv. Funct. Mater., 2026, e31264.
J. Am. Chem. Soc., 2025, 147, 9229.
Angew. Chem. Int. Ed., 2025, e202503568
Chem. Commun.,2024, 60, 2629-2632
J. Am. Chem. Soc., 2023, 145 7569-7579
Angew. Chem. Int. Ed., 2022, e202204851.